
Stroke
An in vitro model for ischemic stroke was developed to measure the cell viability and mortality of rat neurons at different time durations after a three hour period of stroke. The measurements were done with three different assays based on cell cytotoxicity, viability and proliferation. The second part was focused on measuring the extent of protection of the neurons through the two cytokines SDF-1α and BDNF.
Stroke is the second most common cause of death worldwide, a major source of morbidity, and ranks first among causes for long-term disabilities in adults [1].
Stroke is a devastating medical condition, where failure to supply oxygen and glucose to brain cells, leads to their death. Stroke is classified as being either hemorrhagic or ischemic, depending on the underlying pathophysiological process responsible. Approximately 80 % of all strokes are ischemic and the rest hemorrhagic. Ischemic strokes are caused by a clot formation in a vessel providing blood to the brain. Hemorrhagic strokes are caused by the rupturing of a blood vessel [2], [3].
There are no standard therapies available for an ischemic stroke, except treatment with tissue plasminogen activator (tPA) and the mechanical removal of the clot within eight hours after the onset of the stroke. Moreover numerous drug clinical trials have reported disappointing results.
TPA can lyse (break-down) the blood clots within a critical time window of 4.5 hours. Most patients are symptomatic at a much later stage, and even if they have symptoms early enough, a rapid workup, meaning brain CT or MRI to rule out a hemorrhagic stroke must be performed before initiation of therapy. Because of the short treatment window and numerous criteria for exclusion (recent heart attacks, pregnancies, bleeding problems etc.) this therapy is only an option for about 7–10 percent of the patients.
After an ischemic stroke, the cells surrounding the infarct zone are heavily affected by inflammation processes, which may cause further cell damage or even cell death [4].
In case of infection or disease, cells release cytokines, which interact with the immune system, leading to a corresponding protective mechanism affecting the area surrounding the injury [5].
The cell line RCA-6, primary fetal rat hypothalamic neuronal cell line from embryonic day 15–16, was used for the study. The reason, why fetal and not adult cells were used, is that adult cells are less flexible and versatile, because they are fully developed and are therefore harder to treat, as compared to fetal cells in culture. Mice and rat cells play a critical role in medical research. Their availability and rate of proliferation is higher than human neuronal primary cells.
The primary goal of the project was to study the physiological parameters of cells after three hours of oxygenglucose- deprivation (OGD) and different durations of reoxygenation. The time in OGD treatment mimicked the situation during an ischemic stroke and the various durations of reoxygenation described the situation of the cells after breaking down the blood clot (lysis). Therefore, this cell culture model represents the corresponding translational approach of pathological conditions of the vascular system in an ischemic stroke.
A secondary goal of the project was to study the influence of the potential neuroprotective cytokines SDF-1α and BDNF on the neurons after hypoxic treatment. This was done to see if they can reduce the negative consequences of the degenerative neuronal cascade released by stroke. The aim was to stop adverse physiological influence after stroke which causes additional impairment of the tissue by neuroprotection. Cytokines can be classified as either anti-inflammatory or pro-inflammatory. Generally, the former are neuroprotective. Their expression is induced within the first few hours after ischemic stroke. Therefore, cytokines may represent attractive and viable targets in future stroke therapy by protecting residual tissue area from getting damaged after a stroke [6].
A control cell culture group was cultivated under normoxic conditions (CON) and a test group cultivated under hypoxic conditions, meaning OGD, for three hours. After the three hours, CON and OGD cells were reoxygenated for various time durations. The physiological impacts on the cell culture (apoptosis, viability and proliferation) after 2 h, 20 h, 26 h and 48 h of reoxygenation were investigated using several assays (investigative procedures). This was done to show how neuronal tissue changes over a period of time after stroke.
The final step was to treat the cell culture with the cytokines SDF-1α and BDNF after three hours of OGD and measure their protective potential on cell viability and mortality, depending upon how long the cells were reoxygenated. They were treated in the same manner (same assays, durations, conditions etc.) as the cells without protective substance, so that an appropriate comparison could be made.
This was primarily done to find out if the two cytokines have any neuroprotective potential, and secondly to determine in which time window of treatment the best results could be obtained.
Stroke is a neurodegenerative disease with a mortality rate of 33 percent. It accounts for 9 percent (4.4 million) of all deaths per year, whereas about 15 million suffer a stroke every year. The lifetime risk of stroke is 1 in 6 for men and 1 in 5 for women, which means every two seconds, someone, somewhere in the world suffers a stroke [7].
Stroke is essentially a preventable disease with known and manageable risk factors. The established modifiable risk factors for stroke include hypertension, diabetes mellitus, smoking and obesity. An unhealthy lifestyle, meaning poor diet, excessive alcohol consumption and the lack of exercise can increase the risk of stroke [8].
Stroke occurs when the blood supply to the brain is disturbed either because of ischemia or hemorrhage (Fig. 1). Ischemic strokes are more prevalent than hemorrhagic ones, making up approximately 80 percent of all cases.
During an ischemic stroke, a blood clot (thrombus) is formed in an artery and therefore blocks the supply of blood to the brain. A hemorrhagic stroke on the other hand occurs when a blood vessel of the brain ruptures, which causes blood to leak into the brain.
Both types of stroke lead to an insufficient amount of oxygen and glucose supplied to the brain [9].
The loss of neural tissue associated with stroke leads to neurophysiological changes in the brain, which trigger a wide range of behavioural impairments.
The loss of neural tissue can lead to a cascade of neuronal events within residual neural tissue after the stroke, summarized as ischemic cascade.


The symptoms of stroke and extent of its damage vary from person to person, depending on which part of the brain gets affected, as well as the degree and the duration of restricted blood supply. The most common signs can be noticed in the face, arms and speech (Fig. 2). Very often, one side of the face or one arm
feels numb and weak. Slurred speech is another symptom of stroke [11].
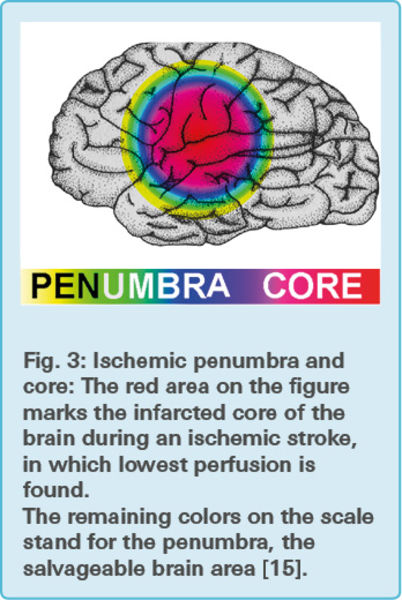
An ischemic stroke is the most common form of stroke. Injury results from tissue anoxia (oxygen depletion) and the lack of glucose, caused by interruption of cerebral blood flow. The ischemic penumbra, an area of constrained blood flow, but structurally intact tissue, is positioned between the lethally infarcted core and the normal brain (Fig. 3). The core has a cerebral blood flow of 20 % less than normal, and cells have lost their membrane potential terminally [13].
Often, within a few hours after the onset of the stroke, the ischemic penumbra is consumed by the progression of the ischemic cascade and becomes part of the damaged core. The typical patient loses 1.9 million neurons per minutes, out of around 100 billion neurons in the brain [14].
An ischemic stroke can be either thrombotic or embolic.
The more common thrombotic stroke often occurs where atherosclerosis in the vessel is already present. A blood clot is more likely to become trapped in a vessel that has been narrowed by plaques. A thrombus then forms inside an artery providing blood to the brain [9].
An embolic stroke results from a blood clot formed in a different body part, which travels through the blood stream and ends up blocking an artery leading to the brain [16].
Thrombus formation involves activation of the platelets (a type of blood cells), as well as the protein fibrin. These two components are normally used to form a blood clot, when a blood vessel is injured. In the case of an ischemic stroke, blood clots are formed even without the blood vessel being injured.
Currently, the only effective therapies for ischemic stroke are an intravenous (IV) injection with tissue plasminogen activator (tPA) and the mechanical removal of the blood clot with a catheterbased device called MERCI.
Cytokines are proteins secreted by their producer cells, which influence the behavior of target cells and sometimes the releasing cell itself. Cytokines are therefore involved in the communication between cells and profoundly affect cell growth and function. They play a crucial role in the immune response and include growth factors, interleukins, interferons, tumor necrosis factors and colony stimulating factors, which means all the proteins controlling growth and differentiation in the human cells [17], [18].
The used cytokine for this project is the recombinant rat SDF-1α (stromal cellderived factor 1- alpha). It is a member of the alpha CXC chemokine protein family, which has the ability to induce chemotaxis. SDF-1α is involved in the development and maturation of the CNS (central nervous system), in neuronal function, in nerve regeneration, and in brain tumors. It plays a central role in hippocampal and cerebellar development. Besides inducing chemotaxis, which is the movement of cells in response to chemical signals, SDF-1α can enhance neuronal cell proliferation and survival. Therefore, it could be a potential neuroprotective substance after stroke, which could minimize the negative outcomes of the ischemic cascade [19].
The second cytokine used for the experiments was recombinant human BDNF (brain- derived neurotrophic factor) a neurotrophic growth factor. Human and rat BDNF sequences are identical, that is why rat BDNF was chosen for the experiments performed on rat neuronal cells. Neurotrophic factors are critical for cell differentiation, neuronal growth, and neuronal survival.
BDNF is found in the brain and the periphery. It has been extensively studied concerning its positive effect on survival promotion and synaptic regulation in the CNS. BDNF positively stimulates neural survival and synapse maintenance by binding to its receptor. It encourages proliferation of existing neurons, as well as growth and differentiation of new ones in the areas vital to learning, memory and deeper thinking. This is why upregulation of BDNF, similar to SDF-1α, forms an attractive approach to treating neurodegenerative diseases like stroke [20], [21].
For this project, the cell line RCA-6, meaning primary fetal rat hypothalamic neuronal cells, of the species Rattus norvegicus – generally referred to as brown rat – was used.
The cells were ordered from the cell bank of F. Hoffmann-La Roche AG. The neurons had already been isolated from the hypothalamus region of the brain of 15–16 days old rat fetuses. The main functions of the hypothalamus region are to produce specific hormones and connect the nervous system to the endocrine system [22].
A primary cell culture is directly derived from the tissue and thus forms a representative model for in vivo studies. Primary cells are very sensitive and differentiate a lot between each other. The used adherent primary neurons had undergone a complex process called transformation, creating chromosomal changes to make them immortal. Adherent cell culture is the system, in which cells are grown as monolayers on an artificial substrate, rather than freefloating in the medium [23], [24].
The reason, why fetal and not adult cells were used, is that adult cells are less flexible and versatile, because they are fully developed and are therefore harder to treat, as compared to fetal cells in culture.
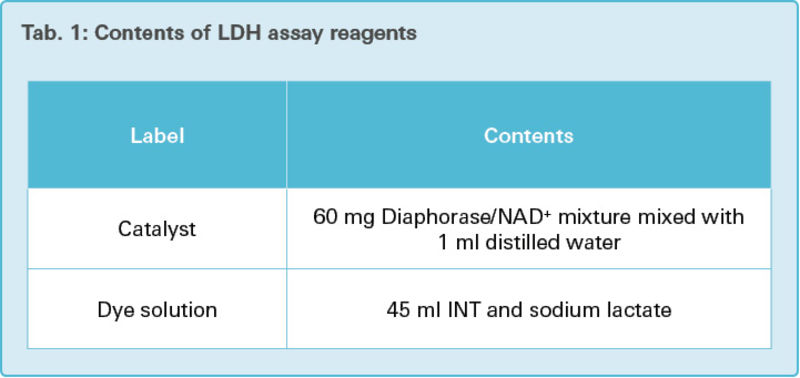
In in vitro cell systems, the cytotoxicity detection assay determines the degree of plasma cell membrane damage, which happens during necrosis. It quantifies the membrane damage by measuring the leakage of intracellular enzyme lactate dehydrogenase (LDH). LDH is a stable soluble cytoplasmic enzyme present in almost all of the cells. LDH is rapidly released into extracellular space when the plasma membrane is damaged and is easy to assay. LDH enzyme activity in the culture supernatant increases during the assay, as does the number of cells with damaged plasma membranes [25].
INT (Iodotetrazolium chloride) is used in this assay to detect the leakage of LDH into the cell culture medium. In the first step, NAD+ is reduced to NADH by LDH-catalyzed conversion of lactate to pyruvate. In the second step, the catalyst (diaphorase) catalyzes the conversion of the tetrazolium salt INT to a colored formazan, by using the newly synthesized NADH. The formazan dye is water-soluble and can be colorimetrically quantified.
The increase in LDH release directly correlates to the amount of formazan formed over time. Because of the linearity of the assay, it can be used to determine the percentage of necrotic cells.
The cell proliferation assay quantifies cellular proliferation, viability, and cytotoxicity with help of the spectrophotometer.
The assay is based on the conversion of the tetrazolium salt MTT into formazan crystals, by metabolically active cells in the cell culture. Enzymes of the endoplasmic reticulum, a type of organelle in the cells, are responsible for the cleavage of MTT to formazan.
The number of viable cells is linearly related to mitochondrial activity. Therefore, the absorption of the formazan dye directly correlates to the number of viable cells [26].
Using fluorescence detection, this assay allows the determination of total cell number, as well as the number of viable and dead cells in parallel. It comprises of three basic steps:
The total number of cells is determined by fluorescence staining of the cell nuclei. Hoechst 33342 is a cell-permeable stain, which can bind to DNA of living and dead cells.
Based on membrane permeability, the percentage of dead cells is determined fluorimetrically. Propodium iodide is a fluorescent dye, which binds to nucleicacids and can only pass through damaged cell membranes.
The amount of viable cells is verified by fluorescence staining using calcein, a fluorescent molecule. The viable dye contains calcein-AM (calceinacetoxymethyl), which is cell membrane permeable, but by itself is not a fluorescent molecule. It is converted to calcein after acetoxymethyl ester hydrolysis by intracellular esterase, an enzyme only found in viable cells [27].
Basically, half of the cell culture plates were deprived of oxygen and glucose (OGD) for 3 hours; they were then reoxygenated for various durations for cell recovery. The assays were performed after specific reoxygenation time had passed, to measure changes in viability, apoptosis and proliferation of RCA-6 cells over a period of time.
Additionally, two cytokines SDF-1α and BDNF were added to the 96-well microtiter plates after OGD treatment.
After the 3 h of OGD treatment the cells were reoxygenated for the same time durations as the cells without cytokine treatment.
The viability, apoptosis and proliferation of the cytokine treated cells were then compared to those of untreated cells.
OGD treatment: For each of the three assays, a part of the cell plates (OGD) was incubated under hypoxic conditions (37°C, 1 % O2, 5 % CO2, 95 % humidity) for three hours. To mimic the glucose deprivation next to the oxygen deprivation in the incubator, the cell culture medium was replaced with glucose-free DMEM for the OGD group.
Normoxic treatment (CON): The other part of the cell plate, serving as a control group (CON) in each assay, was incubated under regular conditions (37 °C, 21 % O2, 5 % CO2, 95 % humidity) with normal glucosecontaining DMEM for three hours.
After the three hours had passed, the cell culture medium was changed to regular growth medium for both the groups (CON and OGD). One part of the cells additionally received either 2 μg/ml SDF- 1α, while another part received 0.02μg/ml BDNF. The concentrations were suggested by the manufacturer Peprotech. The cell cultures were then placed in a regular 21 % O2 incubator, with the amount of time varying between the assays.
To calculate the percentage of cytotoxicity, the following three controls had to be performed in each experimental setup of the LDH:
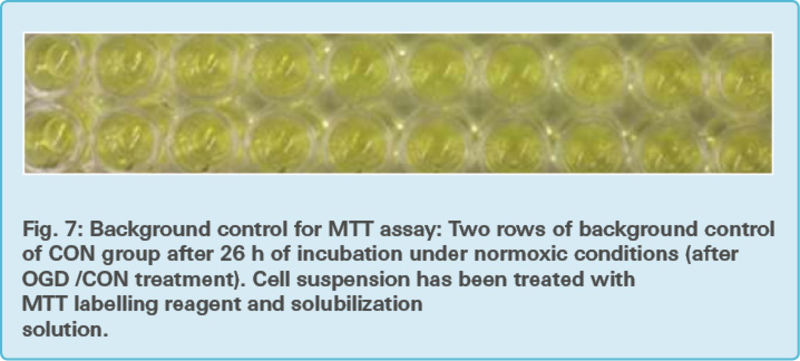
Background control: One part of an additional microplate contained only assay medium without cells and thus the background control provided information about the LDH activity without cells (Fig. 5).
Low control: This portion provided information about the LDH release of normal cells.
High control: By adding 2 % Tween-20 solution to one part of the wells, the high control provided information about the maximum LDH activity in the cells. Tween-20 is a mild detergent that permeabilizes the cell membrane leading to its damage.
Each plate was reoxygenated for 2 h, 20 h, 26 h or 48 h to measure the percentage of necrotic cells over a period of time after OGD treatment.
After the specific reoxygenation time had passed, the supernatant of each well of the 96-well microplate (Fig. 4) was transferred to a new microplate and 100 μl warm mixture of dye solution and catalyst (Tab. 1) was added to the supernatant in each well. The pellets in the remaining plates were used in the MTT assay (3.3). The OGD and CON plates containing the supernatant and added mixture were incubated for 30 minutes at room temperature (24 °C) without light exposure. The absorbance of the samples was measured using an ELISA reader at a wavelength of 490 nm and a reference wavelength of 690 nm, using the software SoftMax Pro. The reference value was subtracted from each reading to normalize the readings, in order to get more accurate data.
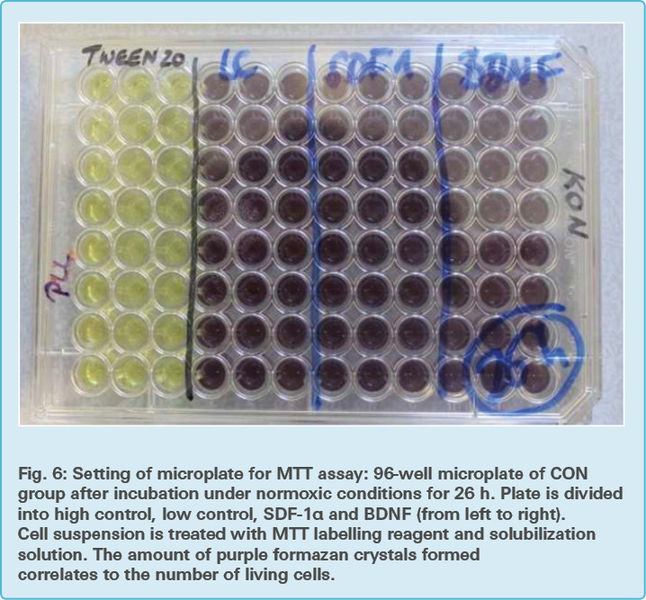
The microplates containing only the pellet from the cytotoxicity detection assay were further used for the MTT assay (Fig. 6 and Fig. 7)
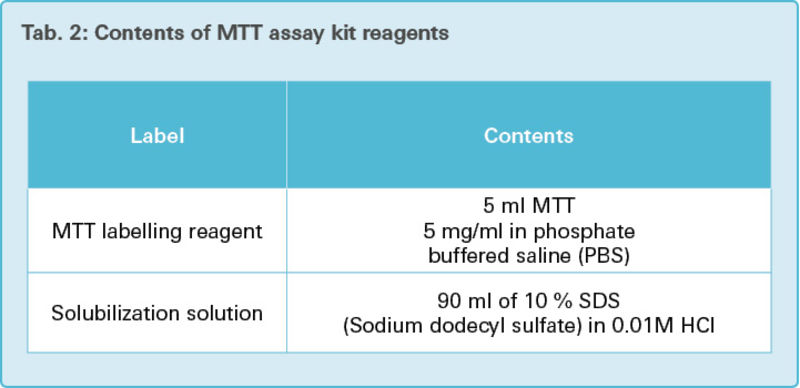
100 μl of growth medium DMEM was added to each well of these microplates. 10 μl of yellow MTT labelling reagent (Tab. 2) was additionally put in each well and incubated in a humidified atmosphere (37 °C, 5 % CO2, 21 % O2, 95 % humidity). After four hours of incubation, 100 μl solubilization solution (Tab. 2) was added to each well and the RCA-6 cells were incubated overnight in a humidified atmosphere.
The microplates were evaluated using an ELISA reader at 550 nm wavelength and a reference wavelength of 690 nm, using the software SoftMax Pro. The reference wavelength served as a normalization value yet again and was subtracted from the rest of the readings.
Cells incubated in the flask were detached using trypsin. They were then counted and 5’000 cells were diluted in 150 μl DMEM per well of four 96-well microplates. No proper images could be obtained using the fluorescence microscope due to the washing away of many cells with this low cell concentration. Hence, the assay was repeated a second time with 7’000 cells in 150 μl medium per well. The microplates were incubated for 24 h in a tissue culture incubator at 37 °C, 21 % O2, 5 % CO2 and 95 % humidity.
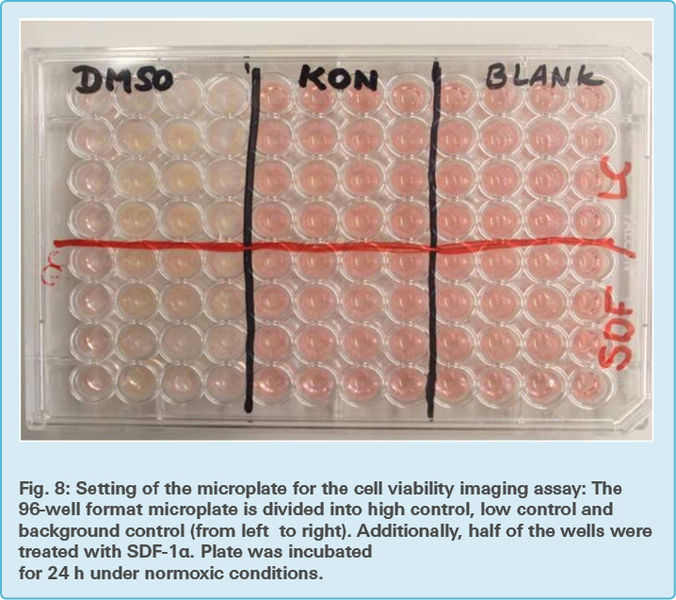
The cell plate was divided into the same three control groups as in the previous assays to obtain comparable results (Fig. 8)
Background control: One third of the 96-well microplate contained only 150 μl DMEM assay medium per well without cells and therefore provided no information about the viability and mortality of the cells.
Low control: One third of the 96- well microplate contained 150 μl assay medium with adherent cells and thus provided information about the normal viability and mortality of the cells.
High control: Due to adding a 2 % concentration of DMSO (Dimethyl sulfoxide) to one third of the wells, those contained only dead cells. The high control therefore provided information about the lowest cell viability. Similar to Tween-20, DMSO permeabilizes the cell membrane and therefore leads to its damage. DMSO is less viscous than Tween-20 and therefore easier to pipet.
The plates were divided into two groups and treated with either OGD or control treatment, as described in chapter 3.2. After this, all of the plates were placed in a normoxic incubator for another 24 h.
The dye mixture was prepared as follows: 25 μl DMSO was added to the viable dye solution (Tab. 3) to make the cells more permeable. For the four microplates, a total of 20 μl nuclei dye was diluted in 20 ml PBS (1:1000 dilution). This mixture was added to the previously prepared mixture of viable dye and DMSO. As the dye is light sensitive, the tube was wrapped in an aluminium foil to protect the dye from light and the bleaching it would cause, which would provide inaccurate results.
100 μl of the dead dye (Tab. 3), as well as 12 μl of the viable dye was added to the tube. After each solution was put in, the mixture was briefly mixed up. 50 μl of this prepared dye mixture was added to each well and mixed with the pipette tip.
After incubating the plates for 30 min in the normoxic incubator, the plates were analyzed using a fluorescence microscope with a 40 fold magnification. The excitation and emission maximum was different for each dye (nuclei, viable, dead) and had to be set for image acquisition.
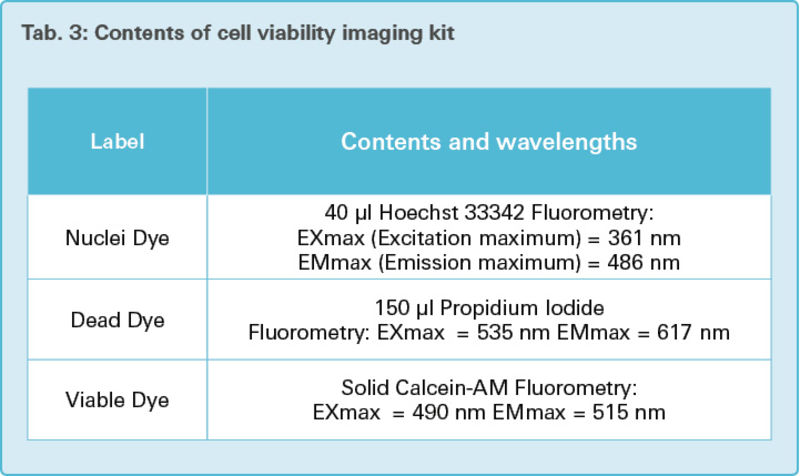
Table 3 lists the contents and fluorometry excitation and emission maximum of reagents used in the cell viability imaging assay.
The main results are:
- The three hour time period in oxygenglucose-deprivation clearly lowered cell viability at every time point.
- Cell viability gradually decreased till about 1 day after blood clot lysis and then increased.
- SDF-1α and BDNF increased cell viability at almost every time point.
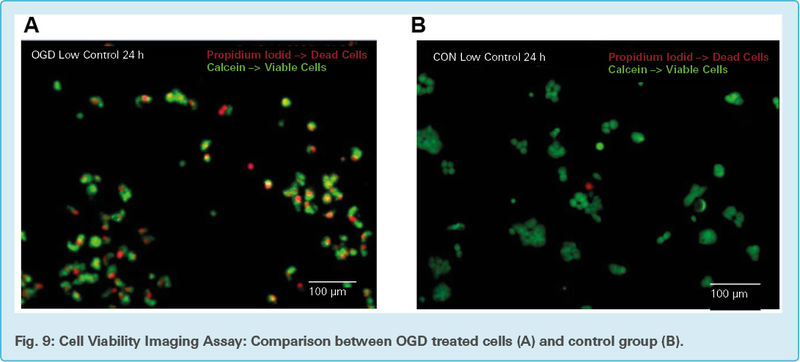
OGD induced changes in cell mortality and viability are visible between OGD treated neurons (Fig. 9a) and untreated neurons (Fig. 9b). Green viable cells are stained with calcein and the red dead cells stained with propidium iodide. The picture was taken with a fluorescence microscope 24 h after the OGD treatment with the scale bar indicating100 μm.
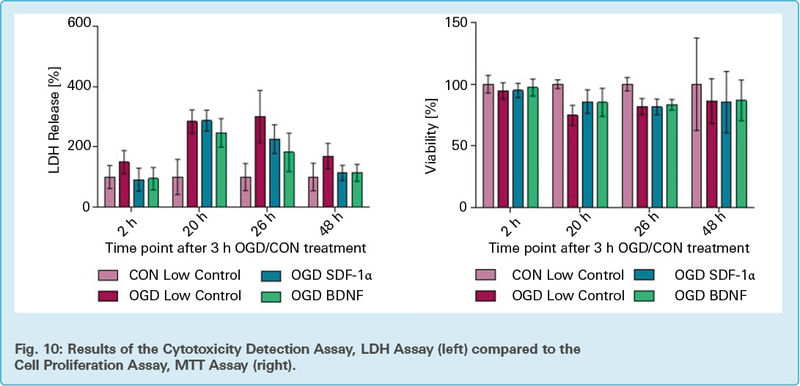
In Fig. 10 the mean values and standard deviation of OGD induced changes in cell mortality and viability of the cells, measured with LDH (A) and MTT (B) assay are shown. Untreated cells (CON low control) and cells treated with OGD, OGD and SDF-1α or OGD and BDNF are compared at different time points after 3 h of OGD/CON treatment. For each trial (n=2 with 100’000 cells/well), 24 technical repeats were measured for each time point (2 h, 20 h, 26 h and 48 h) and their corresponding conditions.
Fig. 11 is an overview of the results of the cytotoxicity detection assay, comparing all of the conditions (described with different colours in the graph legend). It doesn’t focus on any explicit group, but gives an insight into all the groups outlined above for comparative purposes.
On an average, an increasing amount of LDH release and therefore necrotic cells of OGD low control and CON low control can be observed till time point 26 h. From 26 h onwards, the activity decreases, except for the high control values, where it further increases. A clear trend can be observed in the graph.
The bar graph additionally shows the p-value (statistical significance) of the obtained values. Bars, above the columns, indicated with ** and *** mark respective groups which are statistically significant compared to the CON low control bar (100 % ) of the respective time point.
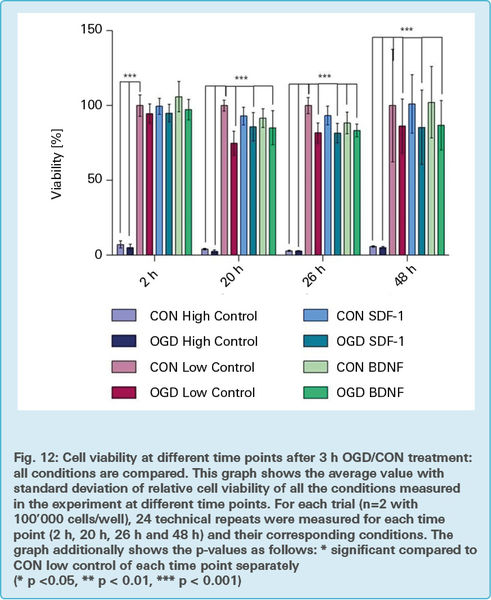
Fig. 12 is an overview of the results of the cell proliferation assay, comparing all of the previously mentioned conditions. It gives a complete picture in one glance, although it doesn’t focus on any explicit groups.
Bars, above the columns, indicated with *** mark respective groups which are statistically significant, as compared to the CON low control (100 % ) bar of the corresponding time points.
The results clearly show a higher mortality rate of neurons after OGD treatment.
The third assay, the cell viability imaging assay, additionally supported the hypothesis of neurodegeneration caused by OGD. The effect of hypoxic treatment was measurable, but not very high, indicating a high stability of the chosen RCA-6 cells.
While the cell viability of OGD low control condition was decreasing up to 26 h of reoxygenation in the LDH assay and up to 20 h after OGD in the MTT assay, it increased suddenly at time point 48 h in both the assays. It can be concluded that the RCA-6 cells take about 2 days to start recovering by themselves after 3 h of OGD. Their lowest viability is reached around 1 day after the stroke, which also shows that the neurons initiated a self-recovery.
After treatment with cytokines, a reduction in cell mortality was measured, compared to the untreated cells. Hence, the hypothesis of the project about the potential neuroprotective effect of cytokines was proven.
It was very hard to draw a precise conclusion about the time dependent effect of the cytokines with the help of cell viability imaging assay, because only major differences were visible through the fluorescence microscope.
The highest cell viability increasing effect of SDF-1α and BDNF varied between the assays and the two cytokines. As per measurements based on both LDH and MTT assays, there was a higher protective effect of BDNF, as compared to the cytokine SDF-1α This indicates that the former cytokine has a stronger protective ability. The highest protection is measured around 1 day after blood clot lysis.
This project being merely the first step in the drug development pipeline, many hurdles need to be overcome and trials done before any proper extrapolation or translation for human patients can be carried out. These trials are very cost intensive. The drug candidates have to pass many tests (especially efficacy in preclinical models, safety, toxicology, side effects, dose response, etc.) prior to commencement of clinical trials in humans. Unfortunately, measuring functional benefit in in vitro models of stroke is difficult due to the lack of behavioural and functional testing and lack of correlation with higher brain functioning in humans. On top of this, there is heterogeneity of stroke in humans, as compared to homogeneous strokes in animals and in vitro cell culture models.
In a nutshell, this project resulted in a successful establishment of an in vitro model to study the pathophysiology of stroke. The study supports the theory about the ischemic cascade released after stroke and suggests that cytokines might lead to an improvement of the condition of stroke patients, by protecting the damaged tissues surrounding the infarct area.
Additional studies based on primary human neurons in a longer time window, and with a combination of different cytokines at different concentrations, would help to understand the real impact of cytokines. The results of the project are consistent with previously published knowledge.
At this point, I would like to express my greatest gratitude to Manuela Jaklin and F. Hoffmann-La Roche AG, who gave me the golden opportunity to carry out this wonderful project in their laboratory. I express my sincere thanks to Dr. Hugo Stocker for his guidance and support.
I would also like to thank Schweizer Jugend Forscht, Biovalley College Network and Taiwan International Science Fair for providing me with a platform to present my project.
Finally, an honourable mention goes to my family and friends for their undivided support and motivation, which provided me with emotional energy required for the successful completion of this project. Without the support of the people mentioned above, this project would certainly not have been successfully completed.
[1] Dirnagl, U. et al., (1999), Pathobiology of ischaemic stroke: an integrated view, In: Trends in neurosciences 22 (9), P. 391–397
[2] Kasner S.E. et al. (2003), Prevention and Treatment of Ischemic Stroke, Blue Books of Practical Neurology, P. 1–5
[3] Bamford, J. et al., (1981), A prospective study of acute cerebrovascular diseases in the community: The Oxfordshire Community Stroke Project, P. 86
[4] Hacke, W. et al., (2008), Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke, New England Journal of Medicine, 359(13): 1317–1329
[5] Meager, Anthony, (1990), Cytokines. Open University Press, Milton Keynes, P. 1–4
[6] Mohr, J.P. et al., (2011), Stroke. Elsevier Mosby. Philadelphia. P. 29-31.
[7] http://www.uhnj.org/stroke/stats.htm (10.12.14)
[8] Interview with Dr. Leo Bonati, the Head of the Stroke Unit of the University Hospital of Basel
[9] Gillen, Glen, (2011), Stroke Rehabilitation: A Function Based Approach, Elsevier Mosby. St. Louis. P. 1–4
[10] (Fig. 1) medicalassessmentonline.com/terms.php (06.12.2014)
[11] http://www.nhs.uk/Conditions/Stroke/Pages/Symptoms.aspx (10.12.2014)
[12] (Fig. 2) Fotolia
[13] http://www.hopkinsmedicine.org/healthlibrary/conditions/nervous_system_s/types_of_stroke_85,P00813/ (10.12.2014)
[14] Brouns, R., De Deyn, P.P., (2009), The complexity of neurobiological processes in acute ischemic stroke, In: Clinical neurology and neurosurgery 111 (6), P. 483–495
[15] (Fig. 3) Dirnagl, U. et al., (1999), Pathobiology of ischaemic stroke: an integrated view, In: Trends in neurosciences 22 (9), P. 391–397
[16] http://www.drugs.com/health-guide/thrombotic-stroke.html (10.12.14)
[17] http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000175/WC500036978.pdf (11.12.2014)
[18] Clemens, M.J., (1991), Cytokines, BIOS Scientific Publishers, Oxford. P. 1, 25,68–72
[19] http://www.peprotech.com/en-US/Pages/Product/400-32A (11.10.2014)
[20] http://www.peprotech.com/en-US/Pages/Product/Recombinant_Human_BDNF/0- (11.10.2014)
[21] Numakawa, Tadahiro (2014): Possible protective action of neurotrophic factors and natural compounds against common neurodegenerative diseases. In: Neural regeneration research 9 (16), P. 1506–1508
[22] http://amrita.vlab.co.in/?sub=3&brch=188&sim=1333&cnt=1 (12.12.2014)
[23] http://www.sigmaaldrich.com/technicaldocuments/protocols/biology/celltypes-culture.html (11.02.2014)
[24] http://www.uta.edu/biology/wilk/classnotes/tissue_culture/Primary%20culture.pdf (11.02.2014)
[25] http://lifescience.roche.com/shop/products/cytotoxicity-detection-kit-ldh- (11.12.14)
[26] http://lifescience.roche.com/shop/products/cell-proliferation-kit-i-mtt- (11.12.2014)
[27] http://lifescience.roche.com/shop/products/cell-viability-imaging-kit-5x96
